We have examined methods to encode sequential data and tested these methods for classifying genes in human DNA. Encoding sequential data is necessary in order to make predictions and it is one of the main topics of Bioinformatics [1][2]. Prior research has shown encoding methods vary in their performance and accuracy [3].
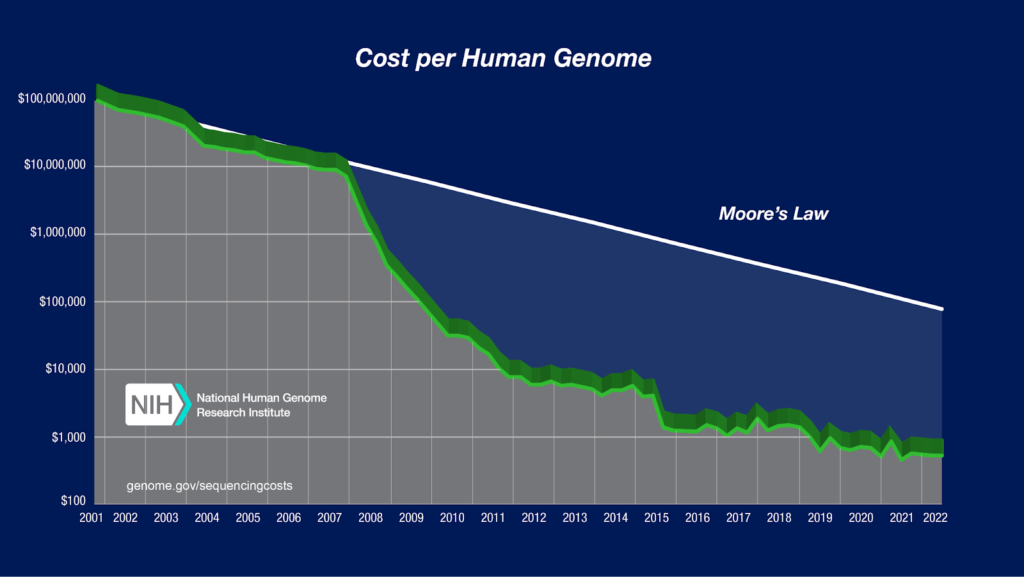
Figure 1. Cost of sequencing per human genome [4]
The amount of DNA data is constantly increasing as sequencing is becoming cheaper. In the future, there will be more need for analyses of large scale DNA datasets, such as population DNA data. The increase in the amount of DNA data creates challenges for pharmaceutical companies and researchers who need to analyze the data.
The first challenge relates to efficient information storage and the second challenge concerns mining useful knowledge from the data [5]. Encoding can solve the first challenge as it can compress data to a smaller space and transmit it faster [6]. Machine learning can be used to solve the second challenge, but compatibility with Machine learning algorithms also generally requires encoding.
Figure 2. Pharmaceutical industry could benefit from improved analyses and tools
One aspect of improving the analysis of DNA data is encoding. Analyses are required to make predictions and understand how medicine is expressed in genes. Researchers and specialists need access to statistical analysis which can improve the diagnostic tools, drug discovery and precision medicine.
Data and Encoding
Nucleotide bases A, C, G, and T are used to express the DNA sequences. A single human genome, that is the complete genetic information of an individual, consists of roughly three billion base pairs, resulting in a file size of approximately 3GB. The dataset we have used is in FASTA form, which is the most common format for sequential data. Although this format is understood by humans, it must be encoded to be machine-readable.
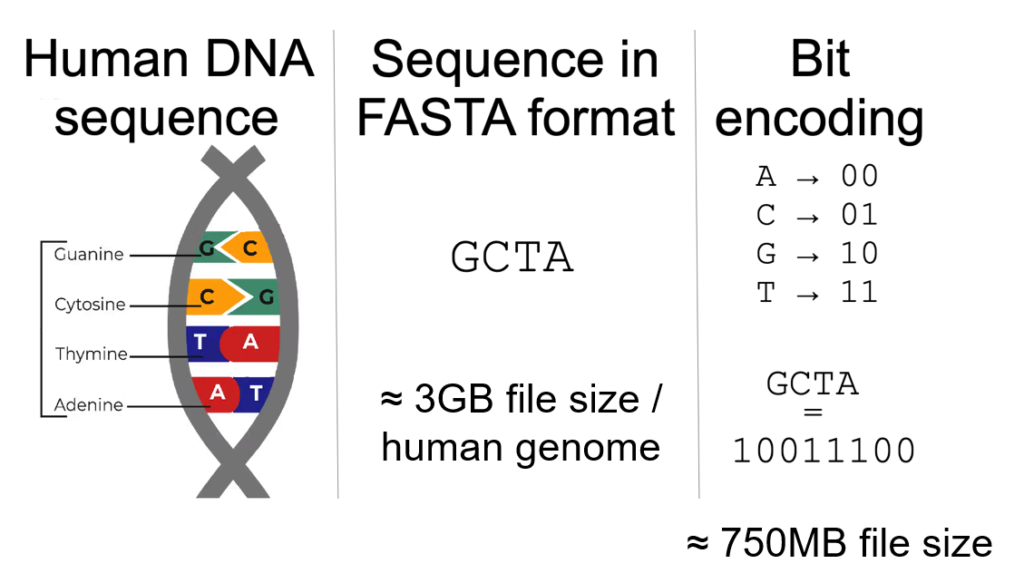
Figure 3. Example of bit-encoding of a DNA sequence.
The above figure is a simple demonstration of encoding each nucleotide base as a 2-bit representation. A significant advantage of this approach is the reduction of the file size to one-fourth of the original sequence and therefore improved efficiency. Most importantly, numerical and binary values are better suited for machine learning models. Several encoding approaches were explored, and we evaluated the three most common encoding methods [7]:
- Ordinal encoding is similar to bit encoding. Instead of encoding to a 2-bit representation, each nucleotide is encoded to a value between 0 and 1. We assigned the values 0.25, 0.5, 0.75, 1 to A,C,G,T.
- One-hot encodes each nucleotide as a vector of length 4. Each vector contains 0 in all places except one, where the value 1 is assigned. For instance, the sequence ‘ACGT’ can be encoded into [1,0,0,0], [0,1,0,0], [0,0,1,0], [0,0,0,1].
- K-mer encoding decomposes a sequence into overlapping segments of length k. For example, with a segment length of 6, the sequence ‘ATGCATGCA’ is divided into ‘ATGCAT’, ‘TGCATG’, ‘GCATGC’, and ‘CATGCA’. This enables leveraging natural language processing tools, such as the bag-of-words model, to analyze the sequences and identify common patterns among them.
ML performance and the effect of encoding methods
We evaluated the predictive strength of each encoding method in conjunction with three different Machine learning models: Logistic Regression, Naive Bayes and Random Forest. The target variable in our data is the human gene family. For the evaluation of each model’s predictive strength, we used Nested Cross Validation. Performance is measured with macro-average f1-score, which is suitable for evaluating accuracy in a multi-class setting [8].
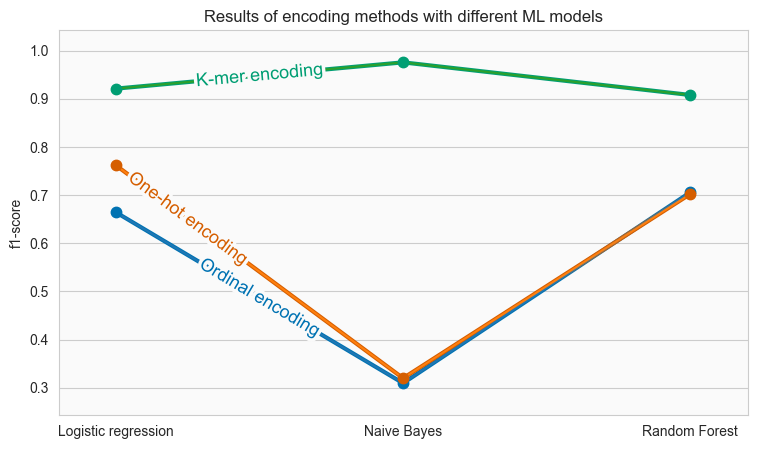
Figure 4. f1 scores for each encoding method and each model. In the x-axis, we have three models and on the y-axis the f1-score. Different colors correspond to three encoding methods.
We notice that K-mer encoding resulted in superior performance for all three models, with f1-scores that exceed 0.9 in all cases. One-hot and ordinal encoding performed well with Logistic Regression and Random Forest, but the f1-scores for Naive Bayes are very low, at just over 0.3.
Inspecting the figure model-wise, we observe that Logistic Regression and Random Forest resulted in f1-score that remained over 0.65. Naive Bayes had the highest diversity.
Overall, when it comes to predictive strength, K-mer encoding has been proven to be the best choice. However, the encoding process of K-mer results in a large number of columns. For this reason, the time complexity of this method was significantly worse than that of ordinal and one-hot encoding. Given that the size of our data was very limited compared to real-life applications, we should keep in mind that K-mer may not be feasible to implement with large datasets.
Use cases
The results are already actionable as they provide recommendations which encoding methods could be used in further analyses. The results can guide the development of multiple applications beyond the scope of our current work.

Figure 5. Example use cases
Use cases could be based on integrating clinical data, involving larger data sets, more sophisticated Machine learning approaches or AI/Deep learning extensions, enabling more comprehensive understanding of different features of the data. Application areas could include analyses of variants in DNA within a population and their role in risk of developing certain diseases, and understanding how medicine is expressed in genes which enables development of personalised medicine.
Introducing the team
Mika Tala is currently working at Acceler8. He has studied Theoretical and Computational Methods at the University of Helsinki.
Jusa Mankki is currently studying at the Data Science Master program at the University of Helsinki and has experience in healthcare software development.
Panagiotis Anastasakis is currently a student of the Data Science Master program at the University of Helsinki. His interests include Bayesian Statistics, Machine Learning and their applications in the field of medicine.
References
[1] Jurtz, V. I., Johansen, A. R., Nielsen, M., Almagro Armenteros, J. J., Nielsen, H., Sønderby, C. K., … & Sønderby, S. K. (2017). An introduction to deep learning on biological sequence data: examples and solutions. Bioinformatics, 33(22), 3685-3690.
[2] Li, Y., Huang, C., Ding, L., Li, Z., Pan, Y., & Gao, X. (2019). Deep learning in bioinformatics: Introduction, application, and perspective in the big data era. Methods, 166, 4-21.
[3] Choong, A. C. H., & Lee, N. K. (2017, November). Evaluation of convolutionary neural networks modeling of DNA sequences using ordinal versus one-hot encoding method. In 2017 International Conference on Computer and Drone Applications (IConDA) (pp. 60-65). IEEE.
[4] National Human Genome Research Institute (NHGRI) – Cost per genome data 2022: https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost
[5] Shastry, K. A., & Sanjay, H. A. (2020). Machine learning for bioinformatics. Statistical modelling and machine learning principles for bioinformatics techniques, tools, and applications, 25-39.
[6] Al-Okaily, A., Almarri, B., Al Yami, S., & Huang, C. H. (2017). Toward a better compression for DNA sequences using Huffman encoding. Journal of Computational Biology, 24(4), 280-288.
[7] Yang, A., Zhang, W., Wang, J., Yang, K., Han, Y., & Zhang, L. (2020). Review on the application of machine learning algorithms in the sequence data mining of DNA. Frontiers in Bioengineering and Biotechnology, 8, 1032.
[8] Opitz, J. & Burst, S. (2019). Macro F1 and Macro F1. arXiv:1911.03347 [stat.ML].